Blog 3: Protein Modelling Pt.3 - Thermodynamics and Statistical Mechanics
Introduction
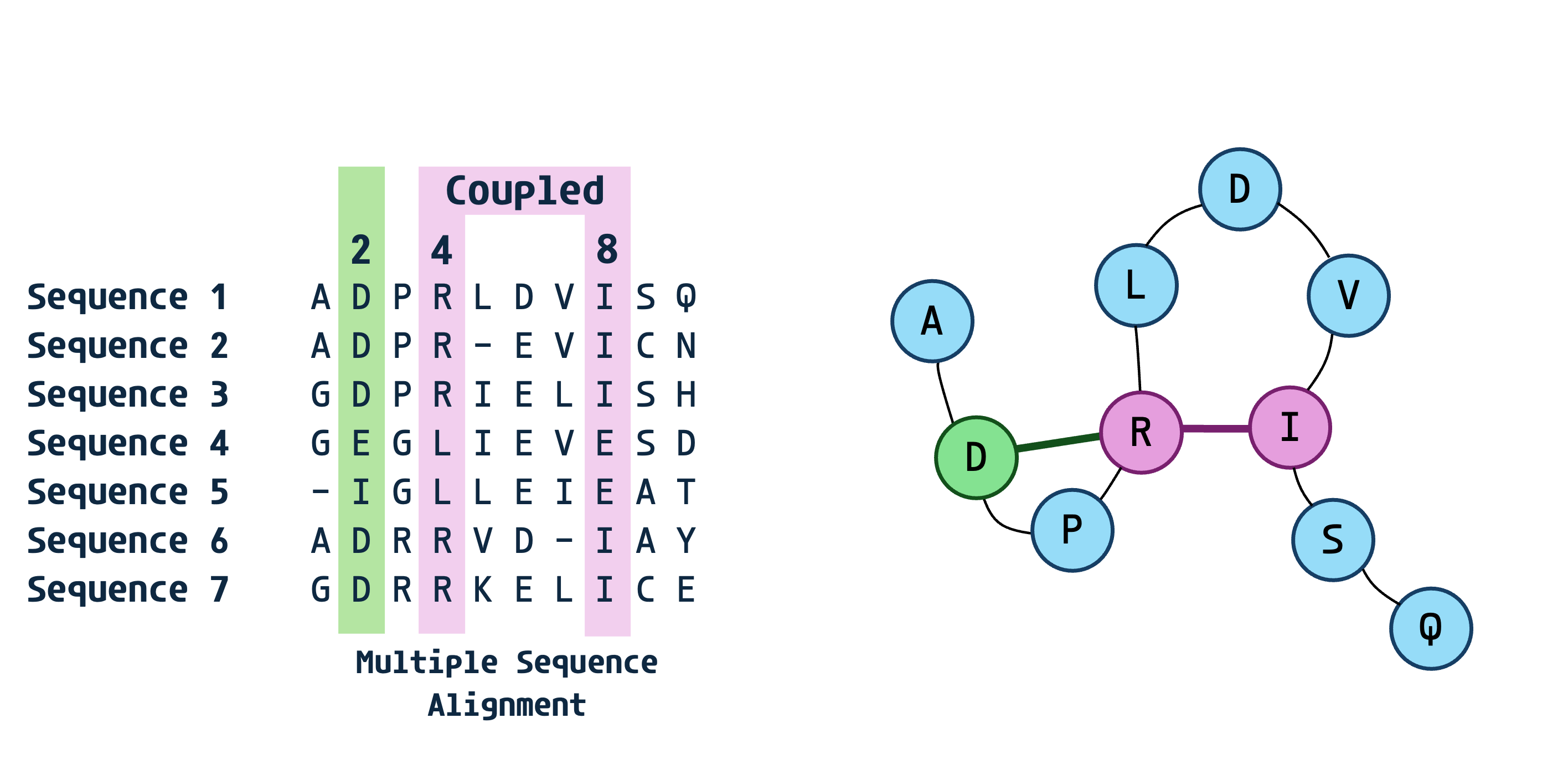
In the previous blog we describe the first method for approximating the Potts model via message passing as proposed by Weigt et al.. The resultant method was somewhat ineffecient relying on a slow iterative belief propagation. In this blog I will walk you through the next iteration in methods for approximating the Potts model, specifically the Mean Field approximation approach pioneered by the same group that introduced message passing. We will walk through the paper by Morcos et al. and they’re more elegant and efficient solution to the Potts model through a marriage of statistical mechanics and thermodynamics to evolutionary sequence analysis.